






以下内容原文发布于AACR官方博客《Cancer Research Catalyst》, 中文内容仅做参考,请点击文末“阅读原文”,阅览原文内容。
数千年前,古希腊人对看似坚不可摧的特洛伊城发动了长达十年的围攻。
了解更多内容,请阅读以下原文。
Dragging Oncogenes From the Cell: A New Horizon for Targeting KRAS
WHAT IS IGA?
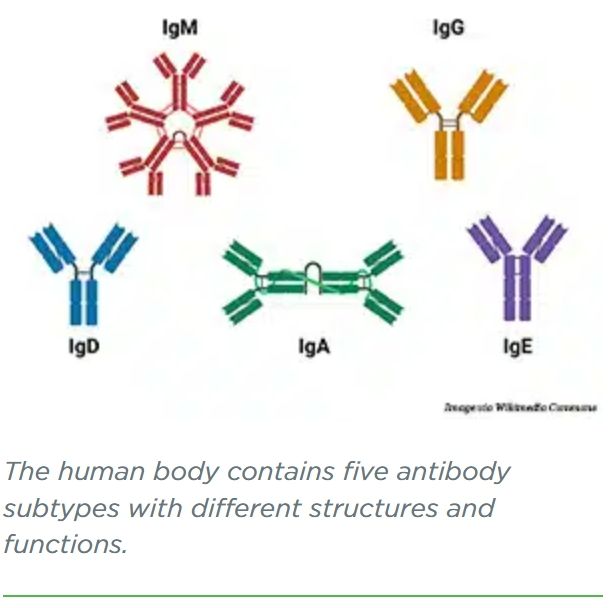
Antibodies are part of the body’s natural defense against infections, foreign substances, and cancer. They identify and bind to potential pathogens, tagging them for destruction by other immune cells. The human immune system uses five main types of antibodies: IgA, IgG, IgD, IgE, and IgM.
A KRAS GRAB-AND-GO
Conejo-Garcia and colleagues set their sights on a notoriously difficult protein to neutralize, the nearly ubiquitous cancer driver KRAS. Mutated in around 20% of solid tumors, KRAS was believed to have an “undruggable” protein surface until the first targeted therapies, sotorasib (Lumakras) and adagrasib (Krazati) were approved by the U.S. Food and Drug Administration (FDA) in 2021 and 2022, respectively.
Sotorasib and adagrasib target the same KRAS mutation, called G12C, which makes up approximately 12% of KRAS mutations across all cancer types and is especially common in lung cancer. However, two more common mutations—G12D and G12V (33% and 23% of all KRAS mutations, respectively)—do not have any FDA-approved targeted therapies. Neither do the various other mutations that can occur in the KRAS gene.
Further, only about one-third of patients with tumors approved for sotorasib treatment respond to the drug, and many who do respond eventually develop resistance.

C
onejo-Garcia and colleagues designed a dIgA antibody that binds to KRASG12D but not to unmutated KRAS, which is found in healthy tissues. Using a cell model of ovarian cancer, the researchers showed that treatment with targeted dIgA decreased the amount of KRASG12D inside the cells and increased the amount outside the cells. When the cells were plated across an impenetrable membrane to divide a well into two chambers, and KRASG12D-targeted dIgA was added to the top chamber, KRASG12D was later found in the bottom chamber, confirming that the migration of dIgA through the cell is directional.
When ovarian cancer cells were xenografted into mice, both intratumoral and intraperitoneal injection of KRASG12D-targeted dIgA decreased tumor growth, while IgG and untargeted dIgA did not. Using data from The Cancer Genome Atlas, Conejo-Garcia and colleagues found that PIGR is highly expressed in virtually all epithelial malignancies (and B-cell lymphoma), indicating that this technique may work in tumor types beyond ovarian cancer.
The researchers tested their targeted dIgA in mouse models of endogenous KRASG12D-driven lung cancer and saw similar tumor shrinkage. In mouse models with intact immune systems, the existence of CD8+ T cells enhanced the antitumor effects of the targeted dIgA, suggesting that the engineered dIgA molecules can stimulate immune responses in addition to neutralizing mutant KRAS.
In both ovarian and lung cancer models, a small-molecule inhibitor of KRASG12D performed similarly to the dIgA, but 20 daily injections were required to produce the same effect as five injections of the targeted dIgA. Conejo-Garcia and colleagues emphasized that the half-life of dIgA (around six days in primates) is significantly longer than most small molecule inhibitors (around six hours), which may help account for the sustained effects.
“We honestly believe that this can change the way that we treat cancer and pave the way for a new generation of immunotherapies,” Conejo-Garcia said during his Annual Meeting presentation.
One aspect of the mechanism underlying the efficacy of KRASG12D-targeted dIgA particularly surprised Conejo-Garcia and colleagues. When dIgA migrates through the cell, it does so enveloped in a cargo vesicle, which should effectively separate dIgA from other cellular contents. However, when the researchers tagged KRASG12D and their dIgA molecules with fluorescent dyes, they found that the two colocalized within the vesicles.
How does dIgA pull mutant KRAS into the vesicles? Conejo-Garcia and colleagues hypothesized that because KRAS exists close to the cell membrane, it might sometimes get swept into the vesicles alongside the dIgA molecules. Alternatively, other vesicles transporting KRAS back to the cell surface may merge with the ones holding dIgA.
In a follow-up experiment, however, the researchers designed a dIgA specific to mutant IDH1, a protein that exists deep in the cytoplasm. In mouse cell xenograft models of colorectal cancer, treatment with IDH1-targeted dIgA decreased tumor size specifically in tumors harboring IDH1 mutations. How dIgA was able to colocalize with a protein that goes nowhere near the cell membrane remains a mystery, but Conejo-Garcia and colleagues intend to follow up on this question in future experiments.
It also remains to be seen whether tumors will develop resistance to targeted dIgA by downregulating PIGR expression, the researchers noted. They suggested that combining dIgA treatment with small molecule inhibitors may help delay or prevent resistance by targeting the cancer driver through different simultaneous mechanisms. The researchers also stated that the safety of such a therapy would need to be evaluated in humans, but they cited some promising data. Antibody preparations containing IgA and IgG mixtures have been used to treat certain infectious diseases with limited toxicity.
Plenty more research is needed, but in the continuing quest to confront seemingly indomitable cancer drivers, perhaps a sneaky approach from ancient Greece may provide a promising path forward.
更多内容,请点击“阅读原文”
排版编辑:肿瘤资讯-Astrid





 苏公网安备32059002004080号
苏公网安备32059002004080号


