本品主要成份为瑞舒伐他汀钙。
化学名称:吡贝地尔
维莫德吉
重组人干扰素
贝伐珠单抗
本品主要成分是左卡尼汀
本品主要成分为亚甲蓝。
格列本脲
本品主要成分为布洛芬。
卡瑞利珠单抗
华法林钠。
本品主要成分为C1酯酶抑制剂。
银耳多糖
本品主要成份为(醋酸)地塞米松。
氟维司群
盐酸吡多醇。
本品主要成分为氟哌啶醇。
来氟米特
本品主要成分为巴氯芬。
恩美曲妥珠单抗
醋酸环丙孕酮。
咪唑立宾
本品主要成份为氨曲南。
维布妥昔单抗
本品主要成份为甲磺酸氟马替尼。
氟脲苷
本品主要成分为英夫利西单抗。
本品主要成分为盐酸阿那格雷。
血卟啉
本品活性成分为硫酸氢司美替尼。
本品主要成分为麦格司他。
依那西普
本品主要成分为妥布霉素。
卡培他滨
食道平散
薄芝糖肽
本品主要成份为绒促性素。
本品主要成份:醋酸钙
本品主要成分为吡贝地尔。
瑞派替尼
盐酸阿柔比星
活性成份:可伐利单抗
可伐利单抗是一种可与补体蛋白 C5(C5)结合的重组人源化免疫球蛋白 G1(IgG1)单克隆抗体。
去铁酮。
本品主要成分为醋酸兰瑞肽。
地舒单抗
碘[I]美妥昔单抗
来曲唑
本品主要成份为盐酸金刚烷胺。
本品主要成分为司替戊醇。
达尔西利
胶体磷[P]酸铬
尼洛替尼
本品活性成份为磷酸芦可替尼。
本品主要成分为重组凝血因子Ⅶa。
盐酸拓扑替康
金葡菌毒素
依维莫司
本品主要成份:卡马西平
本品主要成分为芬戈莫德。
转移因子
氯化镭[223Ra]
瑞戈非尼
丁苯那嗪
本品主要成份为曲前列尼尔。
甲苯磺酸索拉非尼
人参多糖
吗替麦考酚酯
本品主要成分为丙戊酸钠。
主要成份为特立氟胺。
阿扎司琼
伤寒沙门氏菌培养液纯化的Vi多糖
盐酸表柔比星
万他维主要成分为伊洛前列素。
本品主要成份为盐酸普拉克索。
去甲斑蝥素
复方制剂
盐酸多柔比星
主要成分为洛美他派。
主要成份:拉那利尤单抗
甘磷酰芥
还原型谷胱甘肽
西妥昔单抗
E-氨甲环酸
活性成份为阿加糖酶β
匹多莫德
主要成份为:吡非尼酮
唑来膦酸
本品主要成份为 2-(乙酰氧基)苯甲酸。
本品的主要成分为艾曲泊帕乙醇胺。
胸腺肽
本品主要成份为醋酸奥曲肽。
重组人粒细胞刺激因子
主要成分为富马酸喹硫平。
本品主要成分为盐酸氟奋乃静。
氧化苦参碱
本品主要成分为美沙拉嗪。
曲美替尼
维生素B1。
紫芝多糖粉
本品主要成分为酮康唑。
本品主要成分为利鲁唑。
本品主要成份及其化学名称为:
(Z)-N-[2-(二乙胺基)乙基-5-[(5-氟-2-氧代-1,2-二氢-3H-吲哚-3-亚基)甲基]-2,4-二甲基-3-氨
甲酰-1H-吡咯苹果酸盐
放线菌素D
本品主要成分为甲磺酸雷沙吉兰。
本品主要成份为:阿奇霉素。
博安霉素
本品主要成分为:艾加莫德
本品主要成份为:醋酸泼尼松龙。
盐酸氮芥
本品主要成份为地拉罗司。
替吉奥
碳酸钙
本品是由GC1菌株经液体发酵培养法制得的灵芝属薄树芝[Ganoderma capense(Lloyd )Teng]干燥菌丝体粉末中提取制得的灭菌水溶液。其组分为多糖和多肽。
本片适用于特发性低促性腺激素性性腺功能减退症
本品主要成份: 羟丁酸钠
本品主要成分为左旋多巴。
盐酸胍
甲磺酸多拉司琼
本品活性成份为盐酸伊普可泮。
主要成份:美沙拉秦
本品的主要成分为利奥西呱。
司妥昔单抗
卡莫司汀
醋酸甲羟孕酮
本品主要成分为盐酸金刚烷胺。
本品主要成分为卢非酰胺。
因卡膦酸二钠
达沙替尼
本品主要成份为沙利度胺。
本品主要成分为林伐美卡舍明。
去氧氟尿苷
双环铂
帕洛诺司琼
本品的主要成份为苯妥英钠。
本品主要成分为盐酸半胱胺。
磷酸雌莫司汀
硫酸长春花碱
维莫非尼
阿伦单抗
本品主要成分为干扰素γ-1b。
苹果酸舒尼替尼
重组人粒细胞巨噬细胞集落刺激因子
沙利度胺
苯巴比妥
本品的主要成份为西罗莫司。
昂丹司琼
从健康小牛胸腺中提取的胸腺α
盐酸托泊替康
瑞莫杜林主要成分曲前列尼尔。
(-)-N-(1-苯基异丙基)-N-甲基-N-丙基-氯化胺
利可君
4-羟基苯乙酸;5-羟基吲哚乙酸;苯乙酰谷氨酰胺;多肽;马尿酸
盐酸吡柔比星
米泊美生
本品主要成份为:氯苯唑酸
马蔺子甲素
薏苡仁油甘油三酯
硼替佐米
色甘酸钠
本品主要成分为地夫可特。
生物活性的多肽
本品主要成份为波生坦。
左旋门冬酰胺酶
本品含氯化钾(KCl) 应为标示量的95.0%~105.0%。
本品主要成分为依泊汀-θ。
鲨肝醇
本品主要成分为瑞舒伐他汀钙。
胸腺法新
盐酸精氨酸。
本品主要成分为西妥昔单抗。
本品主要成份及其化学名称为:盐酸雷莫司琼
本品主要成份为西罗莫司。
帕妥珠单抗
醋酸甲羟孕酮。
氯膦酸二钠四水合物
本品主要成分为D-3-巯基-缬氨酸。
活性成份为阿加糖酶β。
本最主要成份为瑞派替尼。
盐酸柔红霉素
本品主要成分为托吡酯。
主要成分为盐酸环丙沙星。
三尖杉酯碱
本品主要活性成份为阿伐替尼
本品主要成份为:甲泼尼龙琥珀酸钠。
长春花碱
本品主要成分为甲状旁腺激素。
氟尿嘧啶
派安普利单抗
本品每20 ml溶液含:活性成分:西妥昔单抗 100 mg其它成分:氯化钠 116.88 mg;甘氨酸 150.14 mg;聚山梨酯80 2 mg;一水合柠楼酸 42.02 mg;氢氧化钠(1M) 调节PH直至55;注射用水 加至20 ml。
主要成份:重组人促卵泡激素α
主要成份为巴氯芬。
本品主要成分为氨己烯酸。
恩替诺特/盐酸厄洛替尼
羟基喜树碱
地舒单抗(RANKL 配体的免疫球蛋白 G2 全人源单克隆抗体)
本品主要成分:美沙拉秦。
本品主要成分为促皮质素。
异环磷酰胺
右丙亚胺
阿那曲唑
本品主要成份是甲氨蝶呤
本品主要成分为艾替班特。
恩曲替尼
安吖啶
甲磺酸阿帕替尼
本品主要成份为加巴喷丁
本品主要成分为安立生坦。
卡莫氟
硫酸培洛霉素
苯丁酸氮芥
本品主要成份为奥卡西平。
本品主要成分为盐酸硫胺素。
己烯雌酚
安西他滨
阿帕他胺
西尼莫德
本品主要成份为托伐普坦。
二甲苯磺酸拉帕替尼
重组人白细胞介素-2
顺铂
定位
本品是由GC1菌株经液体发酵培养法制得的灵芝属薄树芝[Ganoderma capense
(Lloyd )Teng]干燥菌丝体粉末中提取制得的灭菌水溶液。其组分为多糖和多肽。
格拉司琼
重组人粒细胞巨噬细胞刺激因子
培美曲塞二钠
艾思瑞主要成份为:吡非尼酮。
本品的主要成份为盐酸司来吉兰
洛莫司汀
抗Tac单抗
亮丙瑞林
本品活性成份为氨吡啶
本品主要成分为:氯苯唑酸葡胺
大麻隆
碘[I]
纳武利尤单抗
本品主要成分为妥布霉素
本品为主要成分为免疫球蛋白。
盐酸小檗胺
本品的主要成分为叶酸。
白介素
螺内酯
本品主要成分为硫唑嘌呤。
羧甲淀粉钠
本品主要成份为环孢素。
重组人粒细胞-巨噬细胞集落刺激因子
骨化三醇
本品主要成份为来那度胺。
盐酸雷莫司琼
本品主要成分为氨甲环酸。
达拉非尼
本品主要成分为来曲唑。
江西山香药业有限公司
本品主要成分为依达拉奉。
化学名称:奥氮平
本品主要成分为替莫唑胺。
盐酸尼莫司汀
本品主要成份为人单克隆免疫球蛋白G2(IgG2)。
主要成分:依托泊苷。
亚胺醌
本品适用于治疗纯合子家族性高胆固醇血症
本品主要成份为:甲泼尼龙
醋酸奥曲肽
本品主要成份为盐酸罗匹尼罗。
门冬酰胺
赛沃替尼
氨磷汀
本片适用于治疗多发性硬化
活性成份:巴氯芬。
本品主要成分为醋酸锌。
舒格利单抗
重组人粒细胞集落刺激因子
西达本胺
活性成分:伊匹木单抗
化学名称:5-氨基水杨酸
本品主要成分为恩他卡朋。
恩沃利单抗
尿多酸肽
阿贝西利
本品主要成份为甲氨蝶呤。
本品主要成分为去铁酮。
紫杉醇/encequidar
福莫司汀
氟马替尼
本品主要成份为硫唑嘌呤。
本品是由GC1菌株经液体发酵培养法制得的灵芝属薄树芝干燥菌丝体粉末中提取制得的灭菌水溶液。其组分为多糖和多肽。
田参氨基酸
去甲斑蟊酸钠
亚叶酸钙
本品主要成份为青霉胺。
本品主要成分为抗坏血酸钠。
氟他胺
醋酸亮丙瑞林
维奈克拉
芬戈莫德
地塞米松棕榈酸酯
甲苯磺酸拉帕替尼
胸腺a
吉西他滨
本品主要成份为安立生坦
活性成份:恩他卡朋
雷莫司琼
胸腺肽α1
盐酸米托蒽醌
本品主要成份为:乙磺酸尼达尼布
主要活性成分:托珠单抗
司莫司汀
巴利昔单抗
雷替曲塞
活性成分:Elosulfase alfa是利用重组DNA技术由中国仓鼠卵巢(CHO)细胞表达制备的重组人N-乙酰半乳糖胺-6-硫酸酯酶(rhGALNS)。
六甲蜜胺
阿达木单抗
曲普瑞林
本品主要成分为甲泼尼龙琥珀酸钠
本品活性成份:司来帕格
斑蝥酸钠
硼替佐米
帕博利珠单抗
本品主要成份为地拉罗司。
本品主要活性成分为重组利妥昔单抗。
含胸腺素α
本品主要成分为布洛芬。
右雷佐生
本品主要成分为碳酸氢钠
本品主要成份为甲氨蝶呤。
a-甘露聚糖肽
本品主要成分为盐酸胺碘酮。
人血白蛋白
十一酸睾酮。
活性成份:艾曲泊帕乙醇胺。
硫酸羟氯喹
本品各类制剂的主要成份为他克莫司。
培唑帕尼
本品主要成分为格列本脲。
替尼泊苷
本品主要成分为吡非尼酮。
测试
本品活性成份为氯巴占
羟喜树碱
本品主要成分为阿巴西普。
环磷酰胺
本品主要成份地西泮。
盐酸槐定碱
辛伐他汀片
活性成分:奥氮平
本品主要成份为醋酸亮丙瑞林。
本品主要成分为亚甲蓝。
盐酸埃克替尼
泊马度胺
甘氨双唑钠
活性成份:盐酸奥扎莫德
本品化学名称为:羟基脲
本品主要成分为溴吡斯的明。
重组人CD22单克隆抗体
榄香烯
他莫昔芬
本品活性成份:马昔腾坦
羟丁酸钠注射液:本品的主要成分为羟丁酸钠。
阿基仑赛
去甲斑蝥酸钠
来那度胺
本品主要成份为利培酮。
本品主要成分为氘代丁苯那嗪。
纳武利尤单抗/瑞拉利单抗
碘[131I]肿瘤细胞核人鼠嵌合单克隆抗体
哌柏西利
本品主要成份为他克莫司。
本品主要成份重组人干扰素β-1b。
硫鸟嘌呤
亚叶酸钠
阿瑞匹坦
本品主要成份:硫酸锌
本品主要成分为左卡尼汀。
氨鲁米特
A群链球菌
西罗莫司
本品主要成分为癸氟奋乃静。
本品主要成份是托伐普坦。
芦可替尼
蛋白质中95%以上为免疫球蛋白
多西他赛
本品活性成份:马昔腾坦
化学名称:N-2-炔丙基-1(R)--氨基茚甲磺酸盐
氯膦酸二钠
重组人粒细胞刺激因子其来源
盐酸多柔比星脂质体
重组人干扰素。
活性成份:巴氯芬
甲基斑蝥胺
甘露醇和氯化钠
重组人血管内皮抑制素
主要成分为他达拉非。
本品主要成份为:利奥西呱
乙双吗啉
乙磺酸尼达尼布
达雷妥尤单抗
本品主要成分为美曲普汀。
本品主要成分为环磷酰胺。
甘露聚糖肽
本品主要成份为枸橼酸西地那非。
伊尼妥单抗
本品主要成分及其化学名:本品主要成分为达那唑。
本品主要成分为奥氮平。
自新鲜猪脾脏中提取的多肽及核苷酸类物质
本品主要成分为艾地苯醌。
磷
戊酸雌二醇。
活性成份:利司扑兰(risdiplam)
乌骨藤
本品主要成分为拉莫三嗪。
塞瑞替尼
硫酸新霉素。
盐酸伊立替康
本品主要成分为左乙拉西坦。
本品主要成分为醋酸钙。
硫酸长春新碱
本品主要成分为培美曲塞二钠。
活性成份:艾美赛珠单抗
克拉屈滨
本品主要成分为达那唑。
甲氨蝶呤
主要成分为考来烯胺。
抗肿瘤免疫核糖核酸
本品适用于纯合子家族性高胆固醇血症
活性成份:利培酮
本品主要成分为依那西普。
布格替尼
硝卡芥
本片适用于治疗视网膜母细胞瘤
本品主要成份为安立生坦。
本品主要成分为戈利木单抗。
长春瑞滨双酒石酸盐
纳米炭
依西美坦
艾度硫酸酯酶β
本品主要成份伊洛前列素。
瑞维鲁胺
氯化锶[Sr]
吉非替尼
本品主要成分为拉莫三嗪
本品主要成分为丁苯那嗪。
磷酸氟达拉滨
磺巯嘌呤
奥布替尼
本品主要成份为癸氟奋乃静。
本品主要成分为二盐酸沙丙蝶呤。
盐酸伊达比星
油酸多相脂质体
克唑替尼
本品主要成份为阿立哌唑。
本品主要成分为乙氨醇艾曲波帕。
醋酸甲地孕酮
人免疫球蛋白
醋酸戈舍瑞林
本品主要成分为卡马西平。
主要成分为亚甲蓝
甲磺酸阿美替尼
胸腺α
卡铂
司妥昔单抗。
本品主要成份为利鲁唑。
乌苯美司
本品蛋白质中95%以上为免疫球蛋白
达卡巴嗪
本品活性成份为帕米膦酸二钠。
本品主要成份为∶溴吡斯的明
氯氧喹
左亚叶酸钙
尼妥珠单抗
枸橼酸西地那非。
活性成分:阿达木单抗40mg。辅 料:枸橼酸钠;枸橼酸;磷酸二氢钠;磷酸氢二钠;蔗糖;聚山梨酯80;氢氧化钠
伊沙佐米
本品主要成份为左氧氟沙星。
度伐利尤单抗
艾曲泊帕乙醇胺
本品主要成分为米托蒽醌。
健康牛胸腺提取含生物活性的多肽类物质
本品主要成分为甘露醇。
普拉曲沙
本品每片含门冬氨酸钾(C4H6NO4K)158mg和门冬氨酸镁[(C4H6NO4)2Mg]140mg。
本品主要成分为利培酮。
阿魏酸钠
本品主要成分为盐酸普拉克索。
苦参碱
本品是前列环素(PGI2)的衍生物。
活性成分:重组人类酸性 α-葡萄糖苷酶
硫唑嘌呤
本品主要成份为加巴喷丁。
尼拉帕利
本品化学名称为:硫酸长春新碱。
肠毒素C
本品主要成分为色甘酸钠。
本品主要成分为培维索孟。
醋酸曲普瑞林
本品活性成份为泽布替尼
活性成分:拉罗尼酶
盐酸博莱霉素
本品主要成份为诺西那生钠。
依托泊苷
本品主要成分为盐酸金刚烷胺。
黄芪多糖
阿托伐他汀钙
盐酸普拉克索
本品主要成分为醋酸亮丙瑞林。
维迪西妥单抗
盐酸博来霉素
本片适用于重症先天性粒细胞缺乏症
化学名称:(+)-(2S)-2-[(4,6-二甲基嘧啶-2-基)氧基]-3-甲氧基-3,3-二苯基丙酸
本品主要成分为吗替麦考酚酯。
多柔比星
盐酸平阳霉素
枸橼酸托瑞米芬
本品主要成分为卡比多巴和左旋多巴。
本品主要成分为司妥昔单抗。
斯鲁利单抗
亚砷酸
盐酸厄洛替尼
本品主要成份为环孢素。
本品主要成分为甲磺酸洛美他派。
羟基脲
卫矛醇
甲磺酸伏美替尼
本品主要成份为氟哌啶醇。
本品主要成分为他达拉非。
美法仑
枸橼酸托烷司琼
阿法替尼
本品主要成份为丙戊酸钠。
本品主要成分为阿达木单抗。
氮甲
α-甘露聚糖肽
维生素B
本品主要成份为苯妥英钠。
醋酸去氨加压素。
盐酸恩沙替尼
辅酶A
奥沙利铂
重组人凝血因子Ⅷ。
本品活性成份为诺西那生钠。
云芝胞内糖肽
本品蛋白质中95%以上为人免疫球蛋白
地西他滨
主要成分托吡酯。
本品主要成份为艾地苯醌。
甲异靛
多磺酸粘多糖
曲妥珠单抗
本品主要成份为海曲泊帕乙醇胺
本品主要成份及化学名称为:醋酸兰瑞肽。
炔雌醇
本品的主要成分为叶酸。
替雷利珠单抗
本品主要成份为更昔洛韦。
本品主要成分为那他珠单抗。
健康牛脾脏为原料
本品主要成分为秋水仙碱。
贝林妥欧单抗
本品主要成份对乙酰氨基酚。
本品主要成份为阿立哌唑
茜草双酯
本品主要成分为甲泼尼龙。
喜树碱
化学名称为:3α,7β-二羟基-5β-胆甾烷-24-酸。
麦考酚酸
本品主要成分为盐酸司来吉兰。
地舒单抗
本品主要成份为:甲硫酸新斯的明。其化学名称为:N,N,N-三甲基-3-[(二甲氨基)甲酰氧基]苯铵甲基硫酸盐。分子式:C13H22N2O6S分子量:334.39
硫酸长春地辛
本品的主要成分为乙磺酸尼达尼布。
本品主要成分为美曲普汀。
福美坦
本品活性成份为盐酸替洛利生。
本品主要成份为伊米苷酶。
高三尖杉酯碱
本品主要成分为去醋酸氨加压素。
酒石酸长春瑞滨
泽贝妥单抗
重组改构人肿瘤坏死因子
本品适用于特发性低促性腺激素性性腺功能减退症
主要成分:酸去氨加压素
本品主要成份为特立氟胺。
亚叶酸钙/盐酸伊立替康/氟尿嘧啶
新福菌素
本品的活性成份为醋酸锌。
本品是由GC1菌株经液体发酵培养法制得的灵芝属薄树芝[Ganoderma capense(Lloyd )Teng]干燥菌丝体粉末中提取制得的灭菌水溶液。其组分为多糖和多肽。
本品主要成分为依库珠单抗。
伊匹木单抗
重组人p53腺病毒
比卡鲁胺
主要成分:达依泊汀 α
本品主要成分为盐酸美西律。
甲磺酸贝福替尼
重组人5型腺病毒
甲磺酸伊马替尼
活性成分:依库珠单抗是采用重组 DNA 技术由鼠源骨髓瘤细胞系 NS0 细胞表达制备的抗人补体蛋白 C5 人源化单克隆抗体(IgG2/4κ)。
辅料: 磷酸二氢钠
磷酸氢二钠
氯化钠
聚山梨酯 80
注射用水
本品主要成份为重组凝血因子Ⅸ。
替加氟
阿立必利
卡博替尼
本品主要成份为左旋多巴。
本品主要成分为托珠单抗。
白消安
槲寄生
凡德他尼
本品主要为替硝苯那嗪
本品的主要成分为马昔腾坦。
甲磺酸奥希替尼
破伤风人免疫球蛋白
环孢素
本品主要成分为奥卡西平。
主要成份为∶吡非尼酮
托烷司琼
健康乳牛(出生24小时内)脾脏为原料提取而成
奈达铂
盐酸沙丙蝶呤。
本品主要成份:盐酸普拉克索。
苦参素
抗人T细胞猪免疫球蛋白
洛铂
主要成分依维莫司。
奥希替尼(osimertinib)
奥拉帕利(olaparib)
阿那曲唑(anastrozole)
曲氟尿苷替匹嘧啶
来曲唑(letrozole)
奈拉替尼(neratinib)
伊立替康(irinotecan)
Tepotinib
卡瑞利珠单抗(camrelizumab)
Ivosidenib
吡咯替尼
LCMC3
贝美替尼(binimetinib)
ibalizumab-uiyk
呋喹替尼(fruquintinib)
尼洛替尼(nilotinib)
雷莫芦单抗(ramucirumab)
卢卡帕利(rucaparib)
平阳霉素
Lurbinectedin
度伐鲁单抗(durvalumab)
trebananib
戈舍瑞林
舒尼替尼(sunitinib)
entrectinib
卡铂他滨(capecitabine)
伊匹单抗
阿帕替尼(apatinib )
tremelimumab
依西美旦( exemestane)
pemigatinib
托瑞米芬(toremifene)
SAR408701
吉西他滨(gemcitabine)
spartalizumab
替雷利珠单抗(tislelizumab)
Infigratinib
阿来替尼
Enfortumab Vedotin
Erdafitinib
艾日布林(eribulin)
仑伐替尼(lenvatinib)
依度沙班(edoxaban)
奥法木单抗(ofatumumab)
测试使用药物标签
氟维司群(fulvestrant)
丝裂霉素
Gardasil 9
布加替尼( brigatinib )
阿法替尼(afatinib)
CAN04(nidanilimab)
阿昔替尼(axitinib)
abivertinib
依维莫司(everolimus )
lorlatinib
硼替佐米(bortezomib)
聚乙二醇化IL-10
福美坦(formestane)
拉罗替尼(larotrectinib)
他莫西芬(tamoxifen)
Trastuzumab deruxtecan
特瑞普利单抗
塞瑞替尼(ceritinib)
奥曲肽
呋喹替尼
Selpercatinib(LOXO-292)
伊布替尼(Ibrutinib)
aldoxorubicin
吡咯替尼(pyrotinib)
YS-ON-001
凡德他尼(vandetanib)
替莫唑胺
alpelisib
恩美曲妥珠单抗(Kadcyla)
S-1
利妥昔单抗
克唑替尼(crizotinib)
Namdenoson
培唑帕尼(pazopanib)
美法仑(melphalan)
泽布替尼
来那度胺(lenalidomide)
达卡巴嗪(dacarbazine)
氨鲁米特(aminoglutethimide)
法米替尼(famitinib)
环磷酰胺(cyclophosphamide)
DS-8201
赞鲁替尼(zanubrutinib)
本妥昔单抗
索凡替尼
紫杉醇脂质体
Mobocertinib (TAK-788)
brentuximab vedotin
曲贝替定(trabectedin)
奈达铂(nedaplatin)
奥拉帕利
larotrectinib
卡博替尼(cabozantinib)
Talazoparib
白蛋白结合型紫杉醇
venetoclax
阿特珠单抗(atezolizumab)
达可替尼
达沙替尼(dasatinib)
Birinapant
索拉非尼(sorafenib )
IBI305
甲氨蝶呤(methotrexate)
奈拉滨(nelarabine)
奥妥珠单抗
维莫非尼( vemurafenib)
PD-1
培美曲赛(pemetrexed)
Brigatinib
拓扑替康(topotecancas)
mirvetuximab soravtansine
信迪利单抗
赛沃替尼(Savolitinib)
本妥昔单抗(Brentuximab)
Zanidatamab
劳拉替尼
Lutathera
曲贝替定(trabectedin)
泊马度胺(pomalidomide)
尼拉帕利(niraparib)
LOXO-101
替西罗莫司(temsirolimus)
T-DM1
Rituximab
idelalisib
伊马替尼(imatinib)
darolutamide
阿柏西普(aflibercept)
SHR-1210
帕妥珠单抗(pertuzumab)
帕尼单抗(panitumumab)
哌柏西利(palbociclib)
PD-L1
阿柔比星(aclarubicin)
阿美替尼
依托泊苷(etoposide)
多纳非尼
王宝成
氟唑帕利
Zanubrutinib
WCLC
伊匹木单抗(ipilimumab)
阿扎胞苷
alisertib
abemaciclib
鲁卡帕尼(rucaparib)
prexasertib
5-FU
卡培他滨(capecitabine)
达拉非尼(dabrafenib)
阿比特龙
吉非替尼( gefitinib)
存达®
贝伐珠单抗(bevacizumab)
苯达莫司汀
奥沙利铂(oxaliplatin)
色瑞替尼
纳武利尤单抗(nivolumab)
阿糖胞苷
多西他赛(docetaxe)
Imfinzi
博来霉素(bleomycin)
阿替利珠单抗
多柔比星( doxorubicin)
tiragolumab
伊布替尼
Bemarituzumab
仑伐替尼
IASLC
恩杂鲁胺(enzalutamide)
卡非佐米(Carfilzomib)
乐伐替尼(lenvatinib)
达卡巴嗪(dacarbazine)
伊马替尼
apalutamide
三氧化二砷
培美曲塞(pemetrexed)
曲美替尼(trametinib)
沃利替尼
厄洛替尼(erlotinib)
neratinib
曲妥珠单抗(trastuzumab)
dinutuximab beta
卡铂(carboplatin)
Savolitinib
瑞戈非尼(ruigefeinipian)
ribociclib
奥西布宁(oxybutynin)
avapritinib
表柔比星(epirubicin)
恩扎卢胺
帕博利珠单抗(pembrolizumab)
氢吗啡酮
西奥罗尼
Dostarlimab
紫杉醇
新辅助
encorafenib
奥拉单抗(Olaratumab)
帕唑帕尼(pazopanib)
二甲双胍(metformin)
埃克替尼(icotinib)
他莫昔芬(tamoxifen)
聚乙二醇脂质体多柔比星
duvelisib
安罗替尼
Durvalumab
拉帕替尼(lapatinib)
avelumab
西妥昔单抗(cetuximab)
CPX-351
顺铂(cisplatin)
巯嘌呤
尼达尼布(nintedanib)







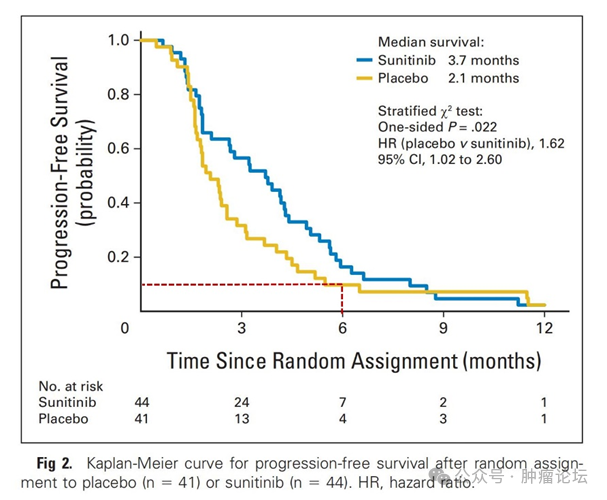


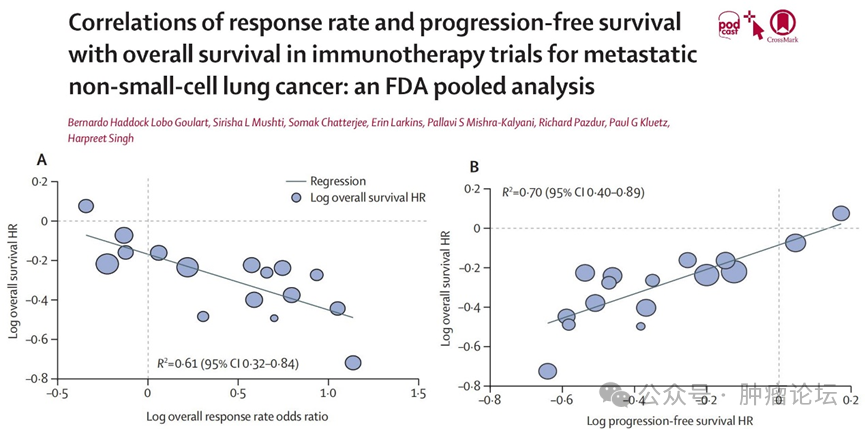

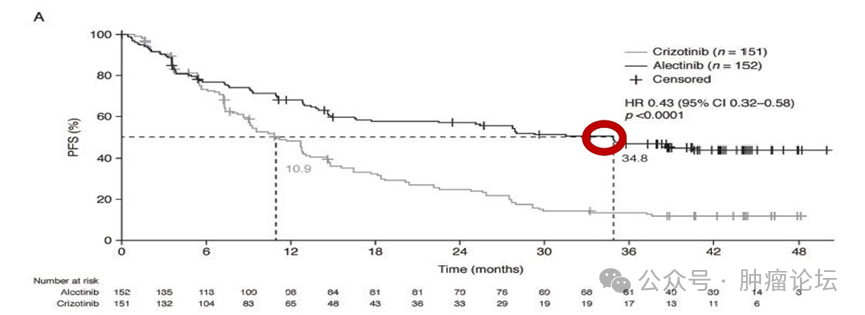
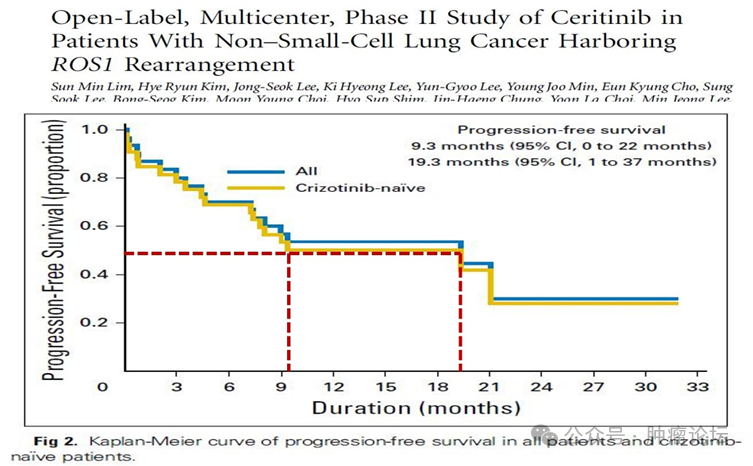





 苏公网安备32059002004080号
苏公网安备32059002004080号


