





当前使用无细胞循环肿瘤DNA (ctDNA)的液体活检在临床研究和实践中都被频繁使用。ctDNA可用于识别可操作的突变,以个性化治疗,检测治愈性治疗后微小残留病灶(MRD),预测治疗的反应。ctDNA也可以从一系列不同的生物体液中分离出来,如果取样距离比血浆近,则有可能检测局部区域MRD,灵敏度更高。然而,在早期和治疗后的MRD环境中,ctDNA检测仍然具有挑战性,因为ctDNA水平极低,导致假阴性结果的风险很高,同时又要考虑克隆造血假阳性结果的风险。为了应对这些挑战,研究人员已经开发出更巧妙的方法来降低ctDNA检测的检测限(LOD),甚至达到百万分之一的范围,并通过减少低水平技术和生物噪声的来源,以及利用ctDNA的特定基因组和表观基因组特征来提高检测的灵敏度和特异性。
自1948年首次发现cfDNA后[1],cfDNA于1977年被发现与恶性肿瘤有关[2]。在肿瘤学领域,美国食品和药物管理局(FDA)于2016年批准的第一个血浆cfDNA检测,即罗氏EGFR突变检测,用于鉴定转移性非小细胞肺癌(NSCLC)患者中EGFR基因的42个突变位点[2,3]。从那时起,大量血浆cfDNA检测进入临床实践,最近很多已开展的液体活检试验主要关注治疗后MRD检测[4,5],以及早期癌症检测,包括结直肠癌、乳腺癌、肺癌、膀胱癌等。该篇综述讨论了cfDNA在识别肿瘤来源的基因组改变中的应用,并描述了ctDNA检测的测序技术范围及其分析的关键方面,还强调了ctDNA在选择靶向治疗、检测疾病复发和MRD、监测治疗反应方面的作用,以及它在免疫肿瘤学中的新作用。
来源与限制
在目前的临床和研究实践中,外周血是ctDNA最常见的来源,收集的体积和储存条件会影响ctDNA检测的敏感性。样品采集和储存方面的创新使血浆样品在室温下保存长达14天而不会出现明显的cfDNA降解,但如果在标准的K2EDTA管中采集血液,则需要更快速的处理。大多数商业cfDNA检测的目标是收集8-20ml全血,产生约4-10ml血浆。
虽然可以对较小的血浆量进行检测,但这些减少的血浆量会影响ctDNA检测的敏感性,特别是在治疗后MRD和早期癌症检测等低疾病负担环境中。随着测序成本的持续下降,辅助治疗选择的扩大,以及ctDNA技术的进步,在治愈性治疗后检测MRD的需求将增加,要求具有高临床敏感性,以准确地为辅助治疗决策提供信息[6]。
检测下限
分析ctDNA最根本的挑战是它的稀缺性,外周血中的大多数cfDNA(90%-99.9%)来自健康宿主,主要是人外周血单核细胞(PBMCs),尽管也来自包括内皮在内的其他健康组织[7-10]。ctDNA的浓度是高度可变的,根据恶性肿瘤类型和肿瘤负荷等因素而有所不同。一般来说,ctDNA在晚期癌症患者中可能占外周血cfDNA的10%,在局部晚期疾病中可能占1%,在早期疾病或治疗后可能占总cfDNA的0.1%[11,12]。
对于晚期癌症患者,较高水平的ctDNA有助于在诊断和治疗耐药时发现临床可操作的突变。然而,对于早期疾病患者或治愈性治疗后的MRD环境中,ctDNA更为有限,使得检测的技术分析更具挑战性。ctDNA检测的分析极限经常根据变异等位基因频率(VAF)进行讨论。实际上,从一根全血采集管(约10ml)的理想外周血中,可以分离出约5ml血浆。在癌症患者中,这种血浆预计含有大约50 ng的DNA (~10 ng/mL),相当于大约15000个单倍体基因组当量[13-16]。在VAF为0.1%的情况下,与局部恶性肿瘤或治疗后MRD一致[17],这相当于只有15个肿瘤DNA分子(图1)。
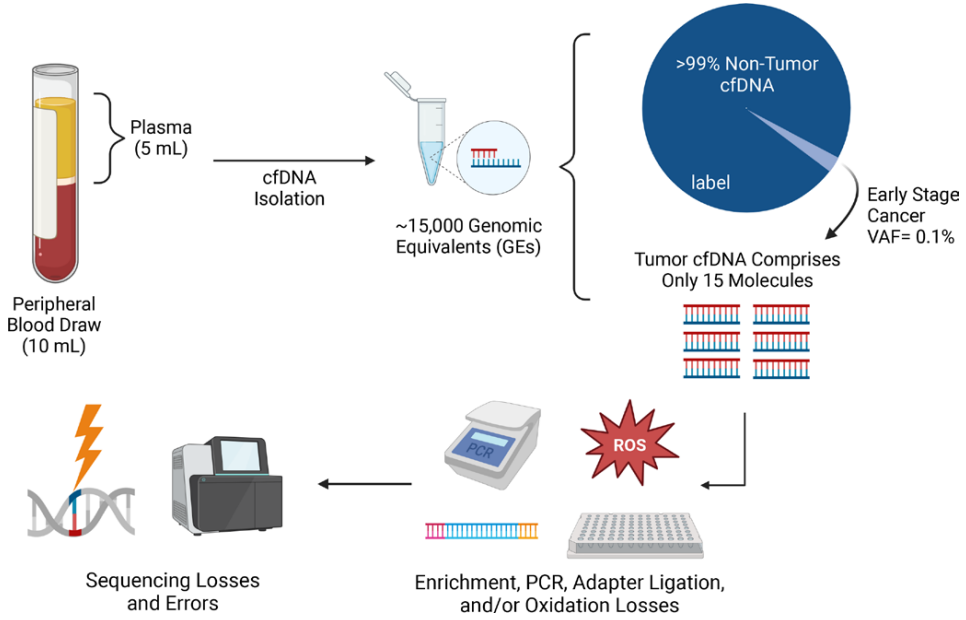 图1 ctDNA检测在早期癌症和MRD环境中的挑战
图1 ctDNA检测在早期癌症和MRD环境中的挑战
由于很少能够将cfDNA样本做到充分测序(在上述示例中,这样做需要独特的测序深度为15000X),因此充分利用到血液样本中具有特定突变的所有15个肿瘤DNA分子将是具有挑战性的。深度覆盖1000X的测序预计只能利用到1个突变分子,假阴性的可能性很高这一问题说明了低VAFs下早期癌症和MRD检测的挑战,在2021年FDA对五种商业ctDNA检测的评估中指出,所有商业分析在VAF水平高于0.5%时表现良好,但在临界值以下就不可靠了,这表明供应商、实验室和分析重复之间的结果不一致。
尽管如此,还是有潜在的解决方案来解决低VAF MRD检测的这些挑战,通过以下方法的组合:(1)肿瘤变异的富集(例如通过片段组学大小选择),(2)个性化测序panel,(3)多突变跟踪,(4)分子条形码区分肿瘤变异与PCR错误,(5)背景错误校正区分肿瘤变异与氧化损伤和非生物学改变。
ctDNA检测方法
PCR
检测ctDNA最直接的方法是PCR。FDA批准的首个罗氏ctDNA-EGFR突变检测就是一种基于RT-PCR的方法。值得注意的是,该检测仅被批准用于没有可用组织进行EGFR测序的已知晚期非小细胞肺癌患者,FDA建议cfDNA检测阴性的患者进行确认性组织检测。该试验分析LOD约为VAF的5%。
数字液滴PCR (ddPCR)是一种提高PCR LOD的替代方法,这是一种微流体技术,于2011年广泛应用,可在油包水液滴内进行单个PCR反应[18,19]。与传统PCR相比,ddPCR和密切相关的技术(BEAMing)的灵敏度提高了10-100倍,研究结果显示VAFs的检测范围从0.1%到0.01%[20,21]。然而,这些技术仍然局限于针对单个或少量已知的突变,这使得它们在早期检测和MRD设置中缺乏灵活性和挑战性,因为检测ctDNA需要同时跟踪多个突变。
PCR方法的局限性促使新一代测序(NGS)技术的发展,以提高灵敏度,降低检测限(LOD),并增加灵活性。杂交捕获和基于多重PCR的NGS都是对传统PCR的重大改进,能够对基因组变异进行更广泛的分析。
基于杂交捕获的NGS和CAPP-Seq
基于杂交捕获的NGS最初是用于细胞DNA的全外显子组测序,然后才适用于cfDNA。在杂交捕获中,确定感兴趣的基因组区域,然后设计生物素连接的互补探针来覆盖这些区域。然后将cfDNA分子连接到条形码接头上,通过PCR扩增,并使用生物素化探针集来“捕获”目标区域。这些探针然后通过结合链霉亲和素包被的磁珠分离,捕获的片段被测序。
CAPP-Seq是一种早期的杂交捕获NGS技术,最初是为分析NSCLC患者的ctDNA而开发的。在最初的CAPP-Seq研究中,针对群体水平肿瘤测序数据以及ALK、ROS1和RET基因的融合和断点区域,设计了一个定制的NSCLC特异性杂交捕获panel,大小约为125 kb,并在人类样本和细胞系中进行了验证,VAFs的检测灵敏度为96%,灵敏度低至~0.02%。
定制的CAPP-Seq panel随后被应用于多种恶性肿瘤,包括弥漫性大B细胞淋巴瘤、食管癌、膀胱癌、前列腺癌、结直肠癌等。杂交捕获NGS方法现在支持FDA批准的两种用于实体恶性肿瘤的ctDNA panel:Guardant360(靶向74个基因,包括snv、indels、扩增和融合)和FoundationOne Liquid CDx(靶向311个基因,包括309个全外显子覆盖)。还首次发现cfDNA的杂交捕获NGS可用于推断外显子组范围内的肿瘤突变负荷(TMB),目前两种商业化的基于杂交捕获NGS检测(FoundationOne Liquid CDx和Guardant360)使临床医生能够无创伤地推断TMB并检测微卫星不稳定性(MSI),这在免疫治疗反应预测中具有重要作用。
在临床实践中,Guardant360和FoundationOne Liquid CDx都被批准作为伴随诊断,帮助确诊实体瘤恶性肿瘤的患者与潜在的治疗方法相匹配。这两个产品在检测snv和indels方面表现最好,对重排的灵敏度较低。例如,FoundationOne Liquid CDx显示,对于可操作的EGFR突变(L858R和外显子19缺失),LOD的中位数约为0.3%,但对于NPM1-ALK融合,LOD仅为~0.9%。因此,FDA对这两种液体活检检测的批准强调,如果可行,ctDNA阴性结果应反映到组织突变检测中。然而,当获得检测结果的时间在临床上很重要或组织活检不可用时,推荐液体优先策略作为组织基因分型的替代选择。
降低LOD以实现更灵敏的MRD检测
原始版本的CAPP-Seq的LOD为~0.02%。随后的CAPP-Seq迭代将这一LOD降低了约10倍至约0.002%,其中包括两项关键创新,即集成数字错误抑制(iDES):分子条形码用于区分真正的突变和PCR错误,背景错误纠正用于抑制文库制备过程中氧化损伤引起的错误。使用iDES增强版本的CAPP-Seq检测局部肺癌患者在治愈性治疗后的MRD,在低至~0.003%的水平下,灵敏度为94%,特异性为100%。
然而,有研究小组报道,使用现代ctDNA检测MRD的灵敏度较低,约为40%[22]。事实上,虽然使用这些方法的ctDNA检测已被证明对MRD检测具有高度特异性,但其敏感性仍然不高,假阴性率可能过高,无法使用当前技术进行稳健的临床实施。这些假阴性ctDNA检测MRD的挑战导致了超灵敏平台的发展,如MRDetect和PhasED-seq,它们采用新颖的策略来进一步降低ctDNA的分析LOD,达到百万分之一的范围。
克隆造血作用
尽管为恶性实体肿瘤开发个性化的肿瘤/正常测序panel可能需要耗费时间和资源,但这种方法已经被许多研究小组采用,以解决生物噪音的关键来源:不确定潜力的克隆造血(CHIP)。CHIP是指造血细胞中与年龄相关的体细胞突变积累,是未来血液恶性肿瘤和心血管疾病的危险因素。如果不仔细处理,CHIP可以显著混淆ctDNA检测并导致假阳性。
当定义为外周血中VAF>2%时,在60岁以上的人群中发现了40%的CHIP突变,随着年龄的增长而增加。当使用较低的VAF阈值和现代测序方法时,低至0.03% VAF的CHIP突变可能包括95%的50-70岁人群,尽管这种低VAF CHIP高流行率的临床意义尚不清楚。CHIP突变通常发生在血液恶性肿瘤相关基因中,典型的有DNMT3A、TET2和ASXL1,也发生在TP53、APC、KRAS、BRCA1和其他一系列与实体瘤相关的基因中,它们也可能由化疗或放疗引起。
一项最全面的ctDNA与CHIP研究比较了124名转移性恶性肿瘤患者和47名健康对照者的cfDNA[23]。所有受试者(肿瘤样本及配对PBMC)都接受了cfDNA中508个基因的靶向超深度测序(60000X)。值得注意的是,配对的PBMC样本中也发现了大多数cfDNA突变(对照组为81.6%,癌症患者为53.2%),与CHIP一致。同样,CCGA对836例实体瘤恶性肿瘤患者和576例对照患者进行了匹配的血浆和PBMCs测序[24],并注意到几乎所有个体都有由CHIP引起的体细胞突变,在较低的VAFs下,CHIP率增加,其中7%的个体在VAF>10%时携带CHIP, 39%在VAF>1%时携带CHIP, 92%在VAF>0.1%时携带CHIP。鉴于这些发现,解决CHIP的最保守方法是通过配对PBMC的深度测序来过滤血浆结果,这对于基于tumor-naïve检测查询低水平ctDNA尤其重要(图2)。
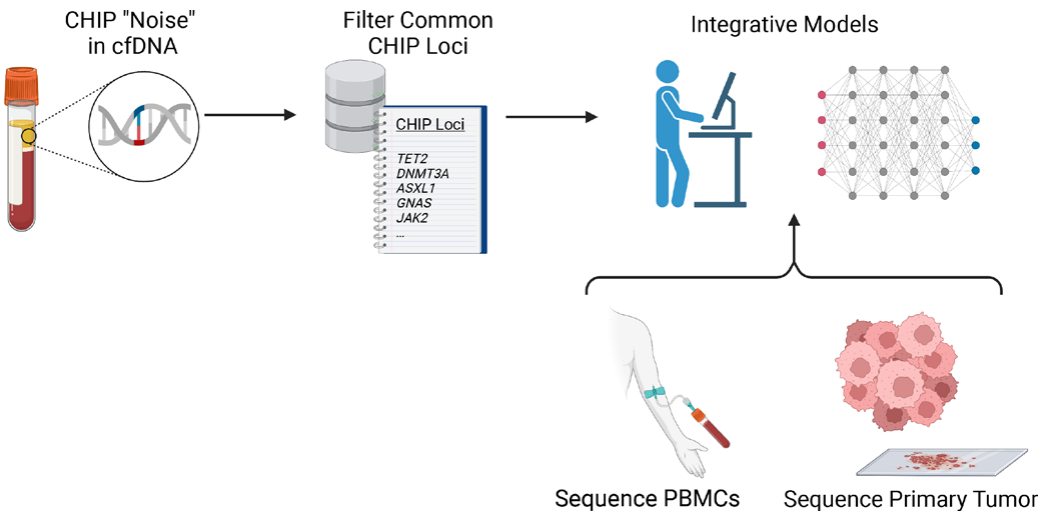
图2 解决克隆造血问题
研究小组还在开发生物信息学方法,试图更可靠地区分CHIP和ctDNA。另一种方法是使用肿瘤信息检测,即追踪在患者肿瘤组织活检中首次发现的血浆变异。由于肿瘤活检通常是在癌症诊断时进行的,而且在手术切除肿瘤后,MRD的问题尤为重要,因此这种个性化的肿瘤知情(tumor-informed)液体活检方法在临床上对许多患者都是可行的,并用于商业化的ctDNA MRD检测,如PCM (Invitae)、RaDaR (Inivata)、Signatera (Natera)和NeXT Personal (Personalis)。
然而,这种肿瘤知情的方法也可能有一些局限性,因为用于设计检测的活检标本可能会遗漏样本组织中存在的亚克隆变异。肿瘤检测也可能受到诊断时获得的活检类型(例如,核心还是细针穿刺)、样本的肿瘤纯度和手术切除标本的质量(例如新辅助治疗的效果)的限制。
新兴技术
cfDNA中基于panel的突变检测,检测灵敏度最终受到每个panel包含的突变位点范围限制,而在复发性突变之外进行更广泛的测序可以提高ctDNA的检测极限,这对早期癌症诊断和MRD检测的新兴应用具有重要意义。这些更广泛测序方法的目标包括甲基化DNA,全基因组拷贝数改变(CNAs)和片段水平测序特征(片段组学)。
最近的研究进一步表明,cfDNA甲基化和片段化的生物学基础是紧密交织在一起的,因此可以从一组特征中推断出另一组特征。最终,cfDNA甲基组学和片段组学都是表观基因组现象的测量方法。除了在早期癌症和MRD检测方面具有转变临床模式的潜力外,这些技术还有望在更基础的生物学研究中促进对癌症表观基因组的更深入了解,并能够通过液体活检无创地跟踪其进化。
cfDNA的替代来源
在低疾病负担环境下,提高ctDNA检测临床敏感性的一种策略可能是对离肿瘤部位更近的非血浆生物区室进行取样。虽然cfDNA和ctDNA通常指的是外周血血浆来源的DNA,但许多研究已经分析了cfDNA,并在不同的生物液体中鉴定了ctDNA,包括尿液、眼泪、唾液、脑脊液、胸膜和腹膜液等(图3)。这些不同的生物区室分别为分析cfDNA的基因组改变带来了机遇和挑战。事实上,从这些替代来源分离的液体可以富集来自局部区域存在的恶性肿瘤的基因组改变片段,这一发现已在粪便(结肠直肠癌)、尿液(尿路上皮癌)、脑脊液(中枢神经系统恶性肿瘤)、以及胸膜和细支气管肺泡灌洗液(肺癌)等中得到证实。与血浆相比,替代生物体液的一个挑战是,它们通常更难以连续收集和处理,尽管唾液和尿液等生物体液比血浆更容易收集,而无需进行静脉切开术。
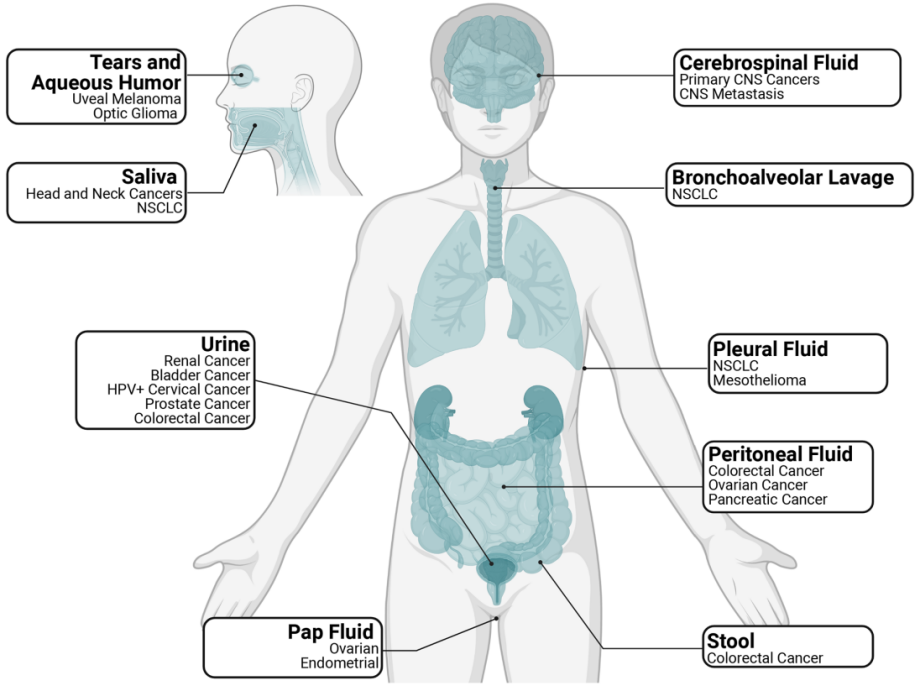 图3 非血浆生物体液和近端相关的恶性肿瘤
图3 非血浆生物体液和近端相关的恶性肿瘤
免疫治疗反应的预测和监测
现代cfDNA液体活检技术的一个新兴目标是预测免疫治疗反应和个性化免疫治疗的管理。TMB是从肿瘤组织测序数据中测量的肿瘤特异性非同义突变的量化,是预测免疫治疗反应的精确生物标志物。TMB也可以通过应用于血浆的杂交捕获cfDNA技术来估计[25],方法是评估设计的靶向panel外显子中的非同义突变的数量。
除了推断TMB,来自Foundation Medicine和Guardant的杂交捕获NGS液体活检分析还包括由短串联重复序列组成的微卫星位点,可以测量MSI,这是免疫治疗反应的另一个重要生物标志物[26,27]。
另一种预测免疫治疗反应的策略是通过ctDNA动力学。具体来说,从免疫治疗前到免疫治疗后,ctDNA水平的变化与免疫治疗反应密切相关,比影像学更早产生评估结果。ctDNA MRD检测也有可能作为根治性手术或放疗后辅助免疫治疗反应的预测性生物标志物。在这方面,Powles等[6]使用Signatera肿瘤知情为基础的NGS检测分析了581例肌层浸润性尿路上皮癌患者的血浆样本,这些患者加入了IMvigor010研究。引人注目的是,手术后但在免疫治疗前可检测到ctDNA MRD的患者在免疫治疗中获得了无病生存期和总生存期的获益,而手术后无法检测到ctDNA MRD的患者则没有。此外,在免疫治疗前可检测到ctDNA但在免疫治疗期间无法检测到的患者,与仍可检测到ctDNA的患者相比,具有更高的无病生存率。Moding等[28]在对局部晚期NSCLC进行明确意向放化疗的回顾性分析中也证明了类似的结果,显示放射治疗后可检测到ctDNA MRD的患者似乎选择性地受益于巩固性免疫治疗,并且在巩固性免疫治疗期间ctDNA水平降低与较长时间无进展相关。
未来方向
ctDNA在临床实践中强有力地检测MRD和预测治疗反应的前景是令人欣喜的,人们继续努力提高ctDNA检测的敏感性和特异性,以识别和跟踪超低频率和罕见的变异,同时考虑背景噪声的抑制。ctDNA分析的前景在不断变化,这里强调的许多技术是在过去几年中发展起来的。最终,ctDNA研究可能会发展到包括综合机器学习模型,例如MRDetec提出的那些克服测序噪声和检测超低频率突变的模型。在本综述的范围之外,还有其他液体分析物,如循环RNA、循环肿瘤细胞、肿瘤诱导血小板和EVs,值得进一步讨论。此外,在ctDNA领域,还有许多其他活跃的研究课题,如亚克隆结构和肿瘤进化的描述、晚期疾病的分子反应、单时间点MRD检测与连续监测的敏感性、不同癌症类型的ctDNA脱落的差异、肿瘤特异性基因组特征如何影响检测和分析策略,以及ctDNA与其他液体活检分析物多组学方法用于早期癌症检测。
虽然人们对ctDNA作为一种肿瘤基因组生物标志物感到兴奋,认为它可以在未来取代标准的影像、病理和实验室检测,但在近期内,能够精确地将ctDNA检测纳入标准诊断模式将是很重要的。对于转移性癌症患者的可操作突变,ctDNA检测已经做到了这一点,指南建议,如果肿瘤组织无法获取或无法满足多个分子标志物检测时,优先推荐血浆检测。同样,对于局部癌症患者的MRD和监测测试,ctDNA分析的结果需要无缝地整合到标准临床实践中,以指导临床决策。对于筛查中的早期癌症检测,检测灵敏度和特异性都需要很高,并且需要明确的临床指南来处理阳性结果,其中还应包括考虑到应对在健康个体中假阳性结果相关的心理社会因素。总之,ctDNA有潜力成为个性化医疗的下一个前沿。
1. MANDEL P, METAIS P. Nuclear acids in human blood plasma. C R Seances Soc Biol Fil 1948;142:241–3.
2. FDA Commissioner . FDA APPROVES first blood test to detect gene mutation associated with non-small cell lung cancer.
3. Roche . Cobas EGFR mutation test V2 package insert.
4. Klein EA, Richards D, Cohn A, et al.. Clinical validation of a targeted methylation-based multi-cancer early detection test using an independent validation set. Ann Oncol 2021;32:1167–77.
5. Cristiano S, Leal A, Phallen J, et al.. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature 2019;570:385–9.
6. Powles T, Assaf ZJ, Davarpanah N, et al.. ctDNA guiding adjuvant Immunotherapy in urothelial carcinoma. Nature 2021;595:432–7.
7. Koh W, Pan W, Gawad C, et al.. Noninvasive in vivo monitoring of tissue-specific global gene expression in humans. Proc Natl Acad Sci USA 2014;111:7361–6.
8. Sun K, Jiang P, Chan KCA, et al.. Plasma DNA tissue mapping by genome-wide methylation sequencing for noninvasive prenatal, cancer, and transplantation assessments. Proc Natl Acad Sci USA 2015;112:E5503–5512.
9. Moss J, Magenheim J, Neiman D, et al.. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat Commun 2018;9:5068.
10. Loyfer N, Magenheim J, Peretz A, et al.. A DNA methylation Atlas of normal human cell types. Nature 2023;613:355–64.
11. Chin R-I, Chen K, Usmani A, et al.. Detection of solid tumor molecular residual disease (MRD) using circulating tumor DNA (ctDNA). Mol Diagn Ther 2019;23:311–31.
12. Heitzer E, Haque IS, Roberts CES, et al.. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat Rev Genet 2019;20:71–88.
13. Newman AM, Lovejoy AF, Klass DM, et al.. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol 2016;34:547–55.
14. Diamandis EP, Fiala C. Can circulating tumor DNA be used for direct and early stage cancer detection? F1000Res 2017;6:2129.
15. Diamandis EP, Fiala C. Can circulating tumor DNA be used for direct and early stage cancer detection? F1000Res 2017;6:2129.
16. Bettegowda C, Sausen M, Leary RJ, et al.. Detection of circulating tumor DNA in Early- and late-stage human malignancies. Sci Transl Med 2014;6:224ra24. 10.
17. Moding EJ, Nabet BY, Alizadeh AA, et al.. Detecting liquid remnants of solid tumors: circulating tumor DNA minimal residual disease. Cancer Discov 2021;11:2968–86.
18. Quan P-L, Sauzade M, Brouzes E. dPCR: A technology review. Sensors (Basel) 2018;18:1271.
19. Hindson BJ, Ness KD, Masquelier DA, et al.. High-throughput droplet Digital PCR system for absolute quantitation of DNA copy number. Anal Chem 2011;83:8604–10.
20. Shields MD, Chen K, Dutcher G, et al.. Making the rounds: exploring the role of circulating tumor DNA (ctDNA) in non-small cell lung cancer. Int J Mol Sci 2022;23:9006.
21. Diehl F, Li M, He Y, et al.. Beaming: single-molecule PCR on microparticles in water-in-oil emulsions. Nat Methods 2006;3:551–9.
22. Pellini B, Chaudhuri AA. Circulating tumor DNA minimal residual disease detection of non-small-cell lung cancer treated with curative intent. J Clin Oncol 2022;40:567–75.
23. Razavi P, Li BT, Brown DN, et al.. High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nat Med 2019;25:1928–37.
24. Swanton C, Venn O, Aravanis A, et al.. Prevalence of Clonal Hematopoiesis of Indeterminate Potential (CHIP) measured by an ultra-sensitive sequencing assay: exploratory analysis of the Circulating Cancer Genome Atlas (CCGA) study. JCO 2018;36:12003. 10.
25. Chaudhuri AA, Chabon JJ, Lovejoy AF, et al.. Early detection of molecular residual disease in localized lung cancer by circulating tumor DNA profiling. Cancer Discov 2017;7:1394–403.
26. Willis J, Lefterova MI, Artyomenko A, et al.. Validation of Microsatellite instability detection using a comprehensive plasma-based genotyping panel. Clin Cancer Res 2019;25:7035–45.
27. Woodhouse R, Li M, Hughes J, et al.. Clinical and analytical validation of Foundationone liquid CDX, a novel 324-Gene cfDNA-based comprehensive genomic profiling assay for cancers of solid tumor origin. PLoS One 2020;15:e0237802.
28. Moding EJ, Liu Y, Nabet BY, et al.. Circulating tumor DNA dynamics predict benefit from consolidation immunotherapy in locally advanced non-small cell lung cancer. Nat Cancer 2020;1:176–83.

排版编辑:肿瘤资讯-Lynn





 苏公网安备32059002004080号
苏公网安备32059002004080号


