





ELK4通过非经典SP1/3依赖性方式激活新血管生成因子LRG1来促进结直肠癌进展
ELK4 Promotes Colorectal Cancer Progression by Activating the Neoangiogenic Factor LRG1 in a Noncanonical SP1/3-Dependent Manner
Advanced Science
PMID:37786278 [IF=14.3]
研究背景
结直肠癌(CRC)是美国癌症相关死亡的第二大常见原因,也是第三大最常见的癌症诊断原因。许多遗传和表观遗传改变导致转录组失调,从而导致CRC肿瘤发生和进展。对CRC中失调转录组的机制理解和对潜在关键失调转录因子的鉴定可能有助于开发新颖有效的靶向治疗方法,并将极大地有益于CRC患者的临床结果。尽管MAPK/MEK/ERK通路在CRC中普遍被激活,但MEK/ERK抑制剂在临床上效果有限。作为MAPK的下游靶标,ELK4被认为主要通过与SRF形成复合物来发挥作用。ELK4是否可以作为CRC潜在的治疗靶点尚不清楚,转录调控机制尚未得到系统分析。
研究方法
研究首先构建Elk4-KO小鼠,在AOM/DSS诱导小鼠结直肠癌模型等各类体内外实验中,证实ELK4促进结直肠癌发生进展的生物学功能。接着通过IP-MS/MS、RNA-seq和ChIP-seq等多组学方法,鉴定出ELK4在结直肠癌中的协同互作转录因子。此外,研究还展开了体内外实验,验证MAPK/ERK通路抑制剂与SP1抑制剂联用是否可以协同抑制CRC的生长。最后,研究基于ELK4-SP1/3转录调控的下游靶基因集,开发了肠癌预后预测模型。
研究结果
为了探索ELK4在CRC中的潜在促肿瘤功能,研究生成了具有ELK4过表达或稳定敲低的HCT116和LoVo细胞。通过CCK8测定确定,ELK4的敲低显著降低了HCT116和LoVo细胞的增殖,而ELK4过表达促进了体外细胞增殖(图1A)。与体外测定结果一致,ELK4敲低抑制了体内肿瘤生长。与对照组肿瘤相比,ELK4敲低的异种移植肿瘤的大小和重量下降,Ki67阳性细胞数量减少(图1B),相反,ELK4过表达促进体内肿瘤生长。细胞迁移测定表明,ELK4过表达增强了HCT116细胞的迁移能力,而ELK4敲低则引起了相反的效果(图1C),在LoVo细胞中也观察到类似的结果。为了探索ELK4在体内的促转移功能,研究建立了两种转移小鼠模型。在注射ELK4过表达细胞的小鼠中,苏木精和伊红(H&E)染色验证了肺和肝脏中肿瘤负荷的显著增加,这表明ELK4的过表达增强了CRC中的肿瘤转移(图1D)。接下来,为了探索ELK4在CRC肿瘤发生中的致癌作用,研究建立了嘧啶甲烷-葡聚糖硫酸钠(AOM-DSS)诱导的野生型和Elk4−/−小鼠结直肠肿瘤发生的模型。与野生型同窝小鼠相比,Elk4−/−小鼠在给予AOM-DSS后结肠肿瘤的数量和大小显著减少(图1E),肿瘤细胞的增殖也显著减少。如图1F所示,来源于Elk4−/−小鼠的类器官生长更慢,离体培养的尺寸更小,类器官数量减少,直径比野生型小鼠的类器官短。这些数据表明ELK4的过表达可以促进CRC中的肿瘤生长和转移。
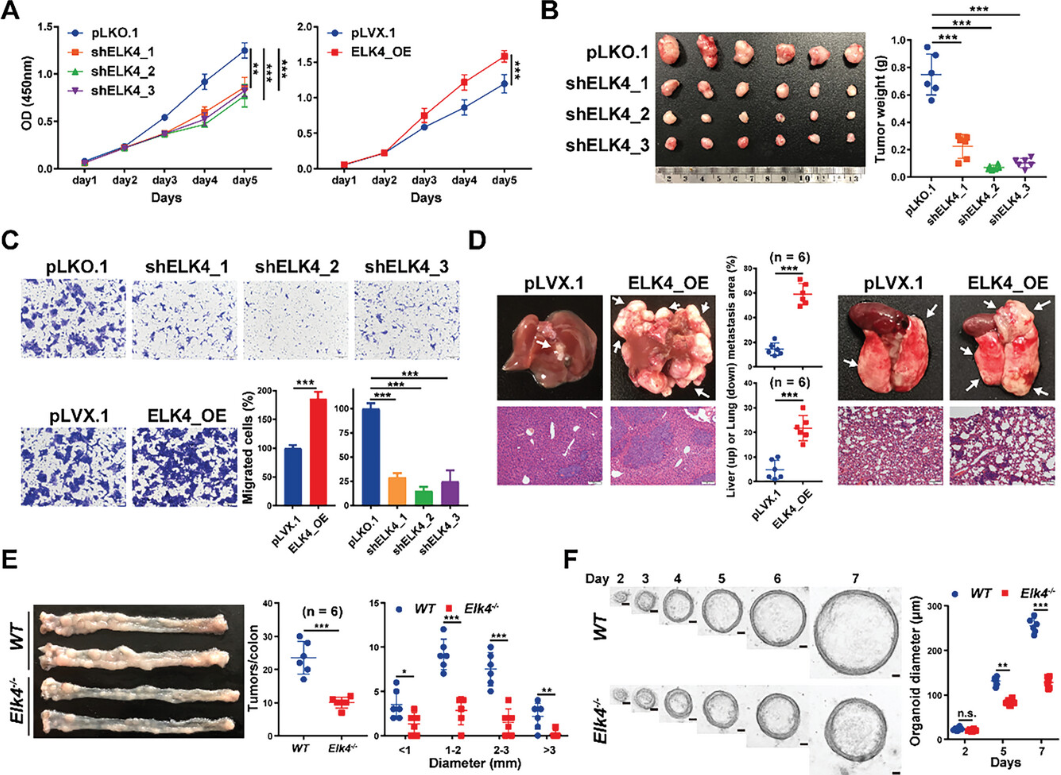
图1 ELK4促进结直肠癌体外和体内恶性生长和转移
为了探索CRC细胞中ELK4的转录靶基因,对ELK4敲低和对照HCT116细胞进行了RNA-seq分析。差异表达基因的基因本体论(GO)富集分析结果显示,前10个富集的生物过程中有两个与血管生成有关(图2A)。差异表达基因富集在“血管生成”中,包括“HOXA3、SRPX2、ID1、CALCRL、FGF1、MCAM和ANGPT2”,以及“血管生成的正调控”,包括“LRG1、THBS1、FGF1、ANGPT2和TWIST1”。研究发现在三种CRC细胞系中LRG1、HOXA3、SRPX2和MCAM下调。除HOXA3、TWIST1和ID1外,其他7个基因都是可由肿瘤细胞表达和分泌的促血管生成基因,这暗示了ELK4在CRC中以旁分泌方式在肿瘤血管生成中的潜在作用。为了验证这一假设,研究使用了体外内皮管形成测定法。来自ELK4过表达细胞的条件培养基(CM)刺激HUVEC的更多管形成,而来自具有ELK4敲低的细胞的CM诱导更少管形成(图2B)。HUVEC的Transwell测定表明,来自ELK4敲低细胞的CM损害了HUVEC的迁移能力,而来自ELK4过表达细胞的CM促进了HUVEC的迁移(图2C)。为了确定ELK4在体内肿瘤血管生成中的作用,研究通过IHC染色检测了ELK4稳定敲低或过表达的CRC细胞形成的异种移植肿瘤中两种肿瘤血管标志物CD31和CD105的表达水平。ELK4的敲除导致异种移植肿瘤中的微血管密度降低(图2D),而ELK4过表达肿瘤中的微血管数量显着增加。此外,对照异种移植肿瘤中肿瘤缺氧标志物HIF1A的蛋白水平高于ELK4敲除异种移植肿瘤中的蛋白水平(图2D),这可能是由于LRG1表达降低下调了HIF1A转录。这反映了ELK4敲除异种移植肿瘤的肿瘤生长减少,这可能是由于肿瘤血管生成减弱。最后,在TCGA CRC数据集中观察到ELK4的表达与CD31或CD34的表达呈正相关(图2E)。这些数据证实了ELK4在CRC肿瘤血管生成中的关键积极作用。
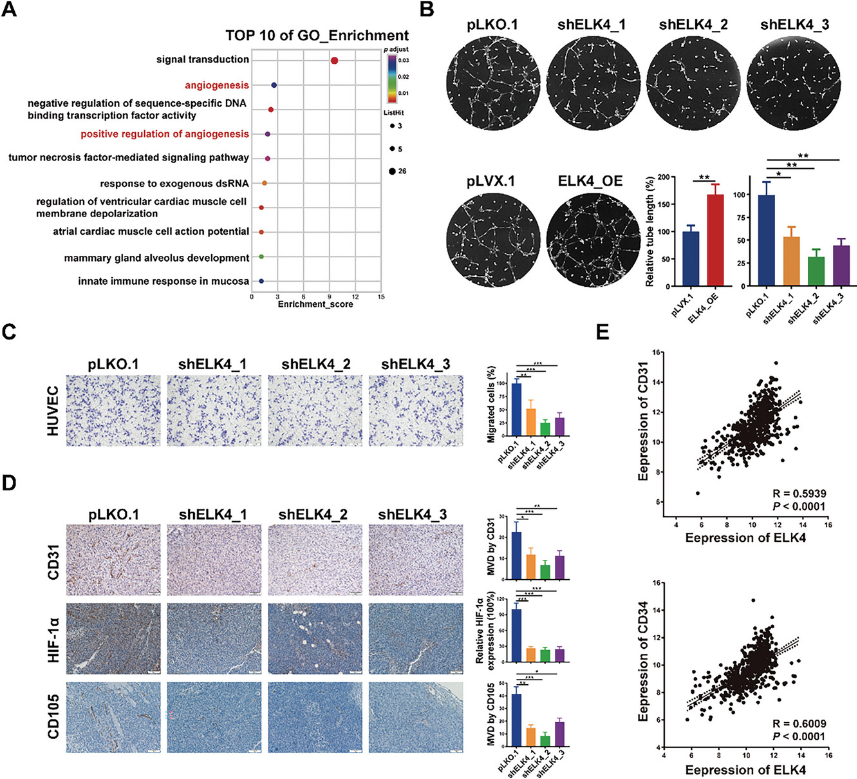
图2 ELK4增强CRC中的肿瘤血管生成
研究还在HCT116细胞中生成了ELK4基因组结合谱,以在全基因组水平上鉴定ELK4的直接靶基因及其相关的转录共调节因子。峰注释显示5685个基因与HCT116细胞中至少一个ELK4结合峰相关。基序富集分析显示,HCT116细胞中的ELK4结合峰高度富集了KLF/SP转录因子家族成员的基序,如SP1、KLF3和SP5(图3A)。研究进行了IP-MS/MS鉴定HCT116细胞中的ELK4结合蛋白,结果发现KLF/SP转录因子家族成员SP1和SP3与HCT116细胞中的ELK4相互作用(图3B)。这些结果提示SP1和SP3可能是CRC中ELK4的关键转录协同调节因子。为了验证该假设,研究获得了已发表的HCT116细胞SP1 ChIP-seq 数据和HEK293细胞中的SP3 ChIP-seq数据。ELK4靶基因集和SP1或SP3相关基因集的交集显示,>85%的ELK4靶基因也被SP1或SP3占据(图3C)。基因组景观分析表明,SP1和SP3在ELK4结合位点周围的区域显著富集(图3D,E)。接下来,研究将ELK4基因组结合谱与HCT116细胞中SRF的公开ChIP-seq数据进行了比较。与基序富集分析的结果一致,大多数ELK4靶标与ELK4结合,但未与SRF结合;大多数SRF靶标与SRF结合,但未与ELK4结合(图3F,G)。这些数据进一步支持了ELK4可能不与CRC细胞中的SRF合作的假设。结合位点相对于基因位置的分类显示,具有共享ELK4-SP1或ELK4-SP3的位点的大部分位于基因启动子中(95.42%),而大多数ELK4、SP1和SP3结合位点通常分布在不同的基因组区域(图3H)。ChIP-seq数据分析进一步提示,ELK4和SP1或SP3占据相同的启动子并形成顺式调节模块以转录调节CRC中的靶基因表达。基因集富集分析(GSEA)也显示SP1和SP3基因特征在ELK4敲低细胞中受到抑制(图3I)。接下来,为了进一步研究致癌转录因子SP1/3在CRC中是否与ELK4有功能上的合作,在稳定过表达ELK4的HCT116和LoVo细胞中敲除了SP1和SP3。研究观察到ELK4过表达诱导的细胞增殖、迁移和血管生成被SP1或SP3敲低显着减弱(图3J-L)。以上研究结果表明,SP1和SP3都可能是CRC中ELK4的关键转录共调节因子,ELK4-SP1/3转录复合物能促进CRC的进展。
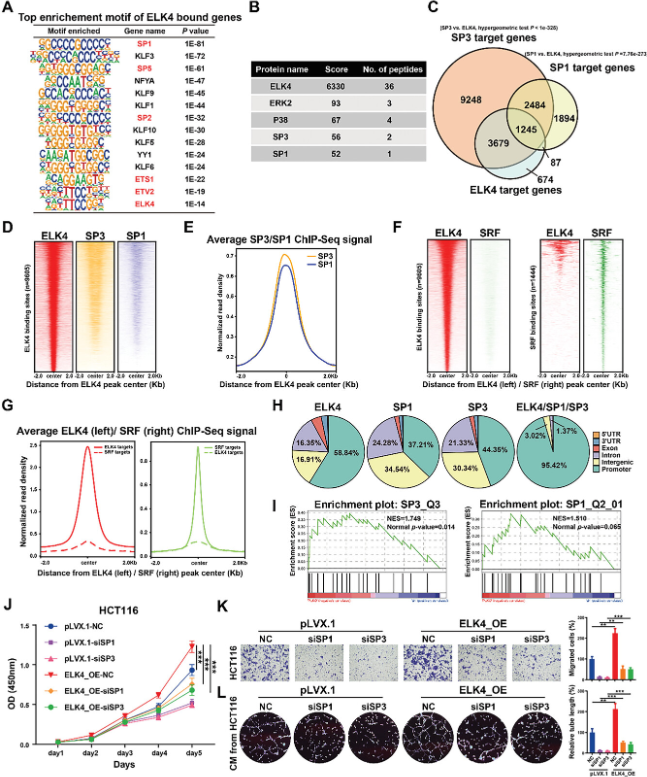
图3 全基因组筛选和功能分析确定SP1/3是CRC中ELK4的关键共调节因子
免疫共沉淀实验证实ELK4可以与SP1和SP3相互作用(图4A)。邻位连接技术(PLA)显示了HCT116、SW480和RKO细胞中内源性ELK4与内源性SP1和SP3之间的相互作用(图4B)。研究试图确定血清诱导的MAPK信号传导是否会影响ELK4和SP1/3之间的相互作用。有趣的是,血清刺激以剂量依赖性方式增强了ELK4和SP1/3之间的相互作用(图4C)。用MEK抑制剂U0126和selumetinib或ERK抑制剂ulixertinib预处理显着减少了ELK4和SP1/3之间血清诱导的相互作用(图4D)。与野生型ELK4相比,P329A突变体ELK4与SP1/3的相关性降低(图4E)。研究发现λ蛋白磷酸酶(λ-PPase)的体外去磷酸化几乎完全消除了血清诱导的ELK4-SP1/3相互作用,这表明磷酸化对ELK4-SP1/3相互作用至关重要。鉴于ELK4在Thr361、Thr366、Ser381和Ser387位点被ERK磷酸化,研究生成了连续的ELK4突变体,其中这四个磷酸化位点被丙氨酸取代。研究表明,T361/T366/S381/S387A突变消除了血清诱导的ELK4和SP1/3之间相互作用的增强(图4F)。研究进一步探讨了ELK4(Thr361、Thr366、Ser381和Ser387)的磷酸化是否会影响其致癌功能。研究发现T361/T366/S381/S387A的突变显着减弱了ELK4在细胞增殖、细胞迁移和肿瘤血管生成中的促肿瘤功能(图4G-I)。综上所述,血清刺激诱导的ELK4磷酸化促进了其与SP1和SP3的相互作用,这可能对ELK4在CRC中的致癌功能至关重要(图4J)。
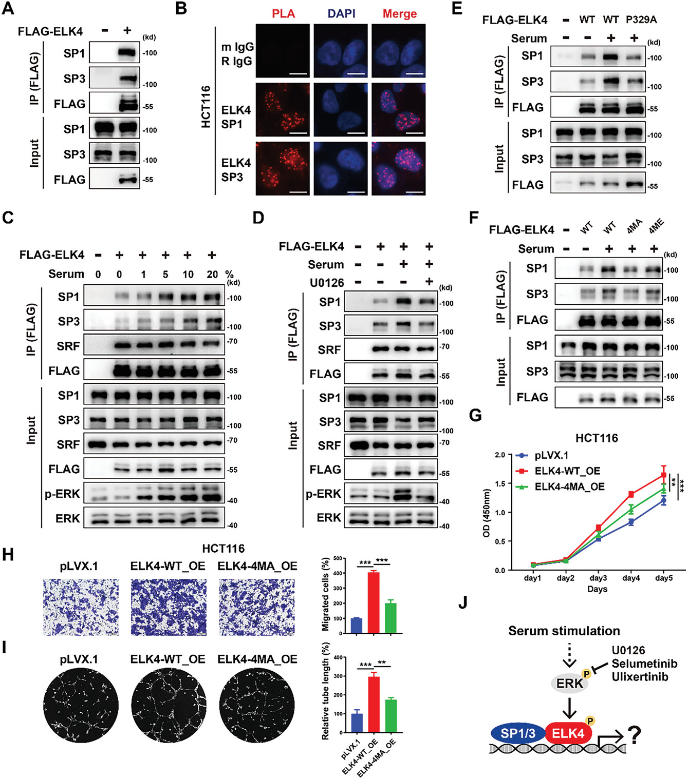
图4 血清刺激介导的ELK4磷酸化促进其与SP1和SP3的相互作用
为了确定受ELK4和SP1/3协同调控以增强肿瘤血管生成的直接靶基因,研究还通过RNA-seq生成了SP1/3敲低HCT116细胞的基因转录谱,并发现通过ELK4敲低或SP1/3敲低差异表达的基因有显着重叠(P < 0.05)(图5A)。LRG1是通过敲低ELK4和SP1/3最强烈下调的血管生成相关基因之一,并且还与ELK4和SP1/3共结合(图5A,G)。研究证实了ELK4或SP1/3敲低后HCT116和LoVo细胞中LRG1的mRNA和蛋白水平降低(图5B、C)。为了进一步验证ELK4-LRG1调控轴在体内的作用,研究检测了野生型小鼠和Elk4-/-小鼠肿瘤组织以及ELK4 OE/KD异种移植肿瘤中LRG1的表达。与体外CRC细胞中的观察结果一致,Elk4-/-小鼠肿瘤中LRG1的mRNA和蛋白水平均有所下降(图5D-F)。ChIP-seq数据显示,ELK4、SP1和SP3占据了位于LRG1基因下游1.4 kb的增强子,这一点在HCT116细胞中通过ChIP-qPCR得到了证实(图5G)。此外,ELK4在LRG1启动子处有一个宽峰,位于LRG1 TSS上游3 kb至4 kb(图5G)。研究还构建了具有ELK4结合位点或SP1/3结合位点突变的增强子+启动子荧光素酶报告基因质粒,结果显示SP1/3截体失去了与ELK4合作共激活LRG1增强子+启动子报告基因的能力(图5H)。此外,ELK4的ERK结合缺陷P329A突变体和ELK4的磷酸化缺陷突变体显示LRG1增强子+启动子报告基因的激活减弱(图5I)。这些数据表明,ELK4和SP1/3之间的相互作用对于ELK4-SP1/3复合体对LRG1的转录激活至关重要。接下来,研究探讨了LRG1是否介导ELK4-SP1/3复合物在CRC中的促血管生成作用。为了阐明这一点,研究将重组LRG1蛋白添加到ELK4敲除HCT116细胞的条件培养基中,观察到重组LRG1蛋白部分减少了管形成刺激的降低(图5J),在过表达LRG1的ELK4敲低HCT116细胞中也观察到类似的结果(图5K)。LRG1的过表达显着减弱了ELK4敲低HCT116异种移植物生长的减少(图5L)。以上研究表明,LRG1受ELK4-SP1/3复合物的转录调控,并且是ELK4-SP1/3复合物致癌功能所必需的。
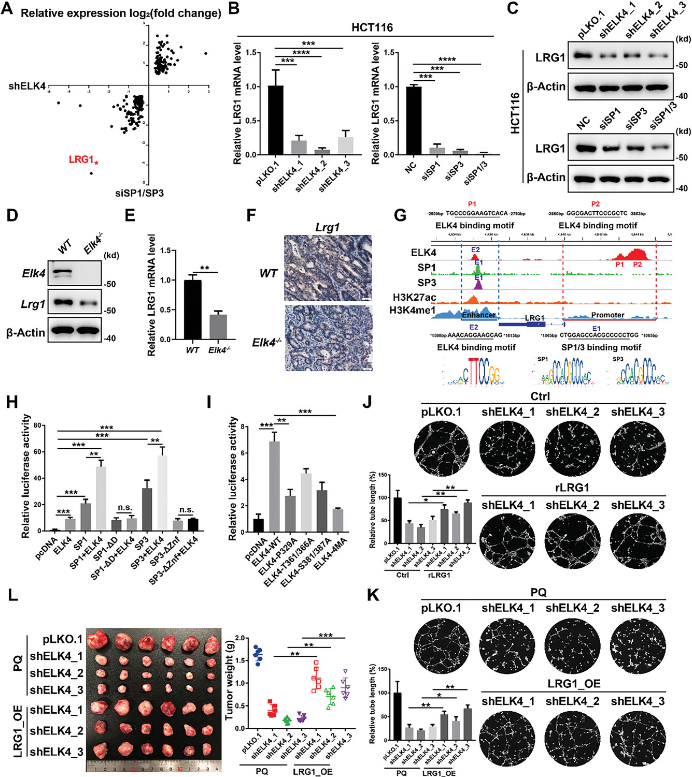
图5 LRG1是CRC中ELK4-SP1/3复合物的直接靶标
以上研究发现ELK4-SP1/3复合物协同调节基因表达以促进CRC肿瘤生长,这提示使用MEK/ERK抑制剂与SP1抑制剂的组合可能对CRC生长产生协同抑制作用的可能。CCK8和克隆形成实验表明,与单一药物治疗相比,CRC细胞与MEK/ERK抑制剂U0126和米曲霉素A(MMA)的共处理对细胞增殖的抑制作用显着增强(图6A、B)。通过Chou-Talalay方法计算的各联合治疗的联合指数小于1,这表明U0126/MMA联合治疗的协同抗肿瘤活性(图6A)。细胞凋亡标志物cleaved PARP1的Western blot分析进一步表明,与单药治疗相比,U0126/MMA联合治疗能诱导更多的细胞凋亡(图6C)。随后,为了确定体内U0126和MMA联合治疗的潜在协同作用,在基于细胞(CDX)和患者来源的异种移植物(PDX)模型中进行了异种移植试验。研究观察到在CDX和PDX模型中,与单一药物治疗相比,U0126和MMA联合治疗显着抑制了肿瘤生长(图6D,E)。为了进一步探索U0126和MMA联合治疗的转化治疗潜力,建立了两个患者来源的类器官(PDO)模型。与异种移植试验的结果一致,单独用U0126或MMA处理导致PDO的生长适度减少,并且联合处理导致PDO的细胞活力显著降低(图6F)。总之,以上数据表明MEK/ERK抑制剂与SP1抑制剂的组合可能是一种很有前途的CRC联合药物治疗。
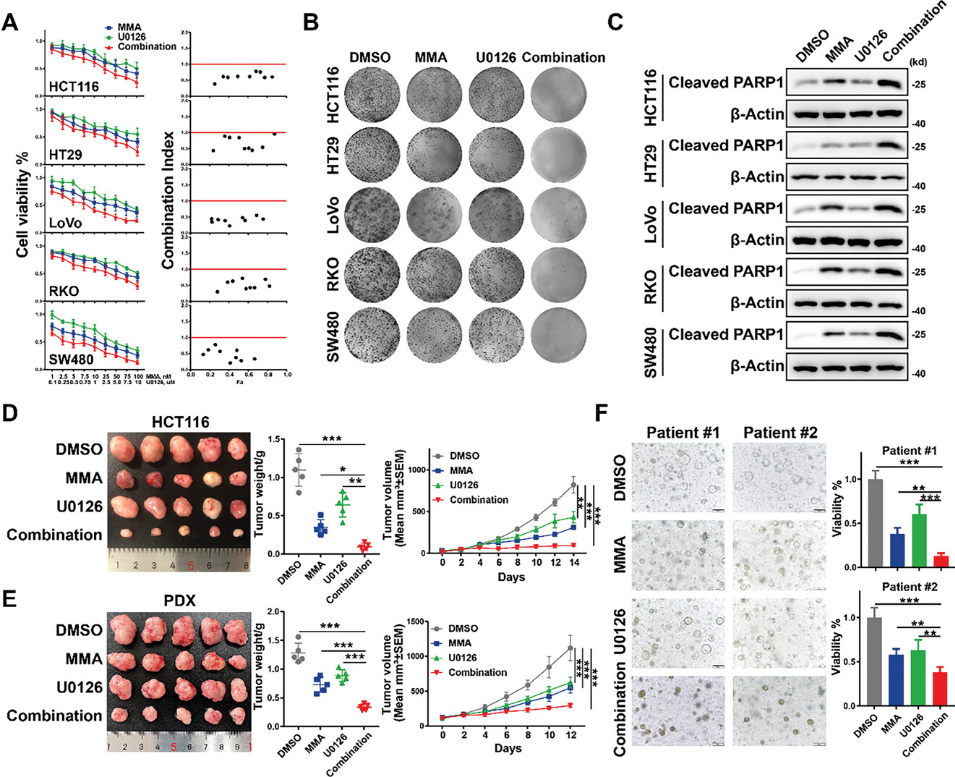
图6 U0126和MMA协同抑制CRC生长
最后,研究还分析了TCGA CRC数据集和GEO数据集(GSE20916)中ELK4的mRNA表达,结果表明ELK4在CRC组织中mRNA表达水平增加(图7A)。研究还在24个CRC组织以及成对的邻近正常组织中验证了ELK4在CRC中的过表达(图7B、C)。对包含190对的CRC和匹配的邻近组织进行了大规模免疫组化分析,结果进一步证实了ELK4在CRC中的过表达(图7D)。此外,Kaplan-Meier分析表明,ELK4表达水平越高,CRC患者的总生存期(OS)和无病生存期(DFS)越差(图7E)。这些数据表明,ELK4的上调是CRC预后不良的一个指标。为了检查ELK4/SP1/3-LRG1轴在CRC中的稳健性,研究在同一CRC队列中进行了IHC染色,评估了SP1、SP3和LRG1蛋白表达。IHC分析显示,LRG1蛋白水平与ELK4、SP1和SP3的蛋白水平呈正相关(图7F)。LRG1和ELK4/SP1/SP3之间mRNA水平的正向关联在TCGA和GSE20916队列中得到进一步验证(图7G)。此外,研究发现归类为ELK4高/SP1高/SP3高的人群显示最差的疾病结局(图7H)。肿瘤脉管系统标志物CD31与CRC队列中LRG1、ELK4、SP1和SP3的蛋白水平呈正相关(图7F)。这些数据表明激活的ELK4/SP1/3-LRG1轴可以促进CRC中的肿瘤血管生成。最后,研究开发了一种基于ELK4-SP1/3复合物调节基因集的基因表达预后模型(图7I)。结果表明基于ELK4-SP1/3复合物调节基因集构建的预后模型可用于独立预测CRC患者的总生存期,进一步支持ELK4-SP1/3转录复合物在CRC中的致癌作用。
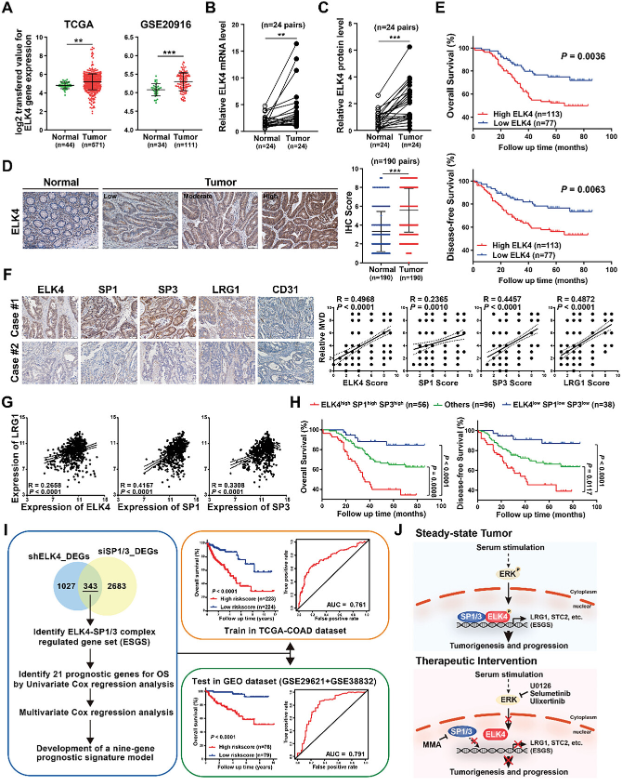
图7 ELK4上调与预后不良的关系以及ELK4/SP1/3-LRG1轴在癌症中的临床意义
讨论与结论
1、ELK4促进CRC肿瘤发生进展。
2、ELK4与SP1和SP3协同作用,转录调节LRG1等,促进肿瘤血管生成、肿瘤生长和转移。
3、血清刺激诱导ELK4的磷酸化,从而促进其与SP1和SP3的相互作用。
4、通过MEK/ERK抑制剂和SP1抑制剂联合治疗靶向ELK4-SP1/3复合物,能够引发协同抗肿瘤作用。
今日评语
该研究通过细致的体内外实验、多组学方法以及生物信息学分析,发现了ELK4与转录因子SP1/SP3互作,以SRF非依赖性方式调控病理性血管新生因子LRG1基因转录,促进结直肠癌发生进展。研究结果表明,MEK/ERK抑制剂和SP1抑制剂联合治疗可提高抗肿瘤活性,这可能是治疗CRC的一种前景广阔的新策略,该研究为结直肠癌的靶向治疗提供了新的作用靶点。当然,在未来仍需要进行更多的研究,开发出更具特异性的SP1抑制剂,以验证这种针对ELK4-SP1/3复合物的治疗策略在CRC中的有效性和安全性。
致谢
浙江大学公共卫生学院 谭玉倩
对以上文章解读做出的贡献
题图来源于网络
【参考文献】
Zhehui Zhu, Yuegui Guo, Yun Liu, et.al. ELK4 Promotes Colorectal Cancer Progression by Activating the Neoangiogenic Factor LRG1 in a Noncanonical SP1/3-Dependent Manner. Advanced Science. 2023 Nov;10(32):e2303378. doi: 10.1002/advs.202303378. Epub 2023 Oct 2. PMID: 37786278
免责声明:本平台旨在分享最新科研资讯,所载内容和意见仅供专业人士参考,不构成任何诊疗建议。在任何情况下,作者及作者所在团队不对任何人因使用本平台中的任何内容所致的任何损失负任何责任。本资料难以设置访问权限,若给您造成不便,还请见谅。若有转载需求,请请在公众号后台留言。
特别声明: 本文属于医学专业文章,仅供医疗专业人员学术交流。不适合作为非专业人士疾病教育或科普用途。
排版编辑:肿瘤资讯-Lisa转载





 苏公网安备32059002004080号
苏公网安备32059002004080号


